INTRODUCTION
Platelets are known to be
engaged in a variety of biochemical and molecular activities designed to
prevent haemorrhage and maintain vascular integrity. To accomplish these tasks, platelets have
surface receptors that can bind adhesive glycoproteins (GP) of various types
and thus promote platelet adhesiveness, aggregation and release reaction. The
platelet activation process involves a number of receptors for agnosits such as
adenosine diphosphate (ADP), epinephrine, thrombin, collagen, fibrinogen,
thromboxame A2, (TXA2) and platelet activating factor (PAF). It also involves
several signal transduction pathways, including phosphoinositide metabolism, arachidonic acid release and
conversion into thromboxame A2, calcium mobilization and phosphorylation of a
number of different target proteins. Many platelet agonists, like thrombin,
ADP, PAF, epinephrine and 5 hydroxytryptamin (5 HT) initiate platelet
activation by binding to transmembrane receptors on platelets coupled with
guanosine triphosphate (GTP) binding proteins (G proteins). The G proteins mediate
a variety of cellular processes by activating different effector molecules,
like adenylyl cyclase, phospholipase C (PLC) or ion channels1.
Epinephrine a very important hormone of the adrenal gland, can influence
platelet aggregation like other agonists. Epinephrine receptors have been
classified as alpha and beta with respective subtypes α 1. α 2,
β1 and β 2. Human platelets contain both α 1, and α 2,
adrenoceptors. The α 2 adrenoceptor is identified as being primarily
responsible for mediating the response to natural agonists2,3. The
receptor (α2) is a G.inhibitory (Gi) protein linked receptor
that contains seven membrane spanning hydrophobic domains, an extracellular
binding site and a series of cytoplasmic binding loops that are the sites of
interaction with Gi proteins in the cytoplasm4. It is also known that activation of α2
adrenergic receptors in human platelets inhibit the adenyl cyclase system
through coupling to a Gi protein5.
It is considered that inhibition of adenyl cyclase system is not sufficient
to cause platelet aggregation, but may be sufficient to amplify the activation
induced by other agonists6.
Indeed, the signal processing mechanisms which mediate the platelet
stimulating effect of epinephrine have not so far been clarified despite
extensive research 7-11.
Calcium ionophore A23187 is
thought to activate cellular phospholipases and thus causes calcium entry into
the cell from the extracellular fluid (ECF). It has also been shown that most
of the agonists act in synergy and potentiate the effect of each other12.
The phenomenon of agonist synergism is very important physiologically and has
been demonstrated in many pairs of agonists13-18. The possible
mechanism of this synergistic action is by raising cytoplalsmic concentration
of Ca++.The first agonist or initial stimulus “primes” the platelets
for an augmented response to a second agonist19. The cytoplasmic Ca++
concentration can be increased by two ways, either by causing an influx of Ca++
into the cell from ECF or by causing a release of Ca++ from
intracellular stores. The role of other effectors and second messengers is not
well understood. The present study was designed to study the potentiation of
epinephrine by A23187 on human platelets and to find out the molecular basis of
this potentiation.
MATERIAL
AND METHODS
This experimental study was conducted on 200 samples
of platelets at Department of Biological Sciences Aga Khan University,
Platelet
aggregation was measured with platelet aggregometer (Model 440, Chronolog
Corporation-USA) using the technique described originally by Born [20]. The
changes in optical density were recorded on omniscribe chart recorder.
Temperature (37Ċ), stirrer speed (1100 r.p.m) and speed of chart recorder
(25 mm per minute) were kept constant. The cuvette containing 500 μl PPP
was placed in PPP reference well. The cuvettes containing PRP were placed in
the incubation wells. At the time of testing each cuvette contained 450 μl
PRP and a Teflon coated stirrer bar. The
final volume of PRP under test was made 500 μl by adding 50 μl of the
test drug. The resulting aggregation
response was recorded for 5 minutes after challenge by the agonists. Aggregation
was induced with epinephrine and A23187 and subthreshold concentration
determined for each agonist. To determine the synergistic effect of epinephrine
and A23187, we added subthreshold concentration of each agonist together. Successive samples of PRP were tested with
different concentrations of drug. Once, the antiplatelet activity of various
inhibitors (Yohimbine, Diltiazem, Verapamil, SNAP) against agonists was
determined, the dose response curves were constructed to calculate IC 50 (Half
maximal inhibitory concentration)values.
RESULTS
The Platelet aggregation induced by different
concentrations of epinephrine (0.1, 0.2, 0.5, 1.0, 5.0 μM) was recorded.
The results indicated 0.2uM as the subthreshold concentration that does not
induce optimal platelet aggregation. The platelet aggregation induced by
different concentrations of A23187 (0.25, 0.50, 1.0, 2.0, 5.0 μM) was also
recorded. The results indicated, 0.5 uM as the subthreshold concentration for
A23187. To establish the synergistic
effect of epinephrine and A23187, the PRP was challenged with subthreshold
concentration of epinephrine (0.2 μM) and subthreshold concentration of
A23187 (0.5 μM) simultaneously and an optimal platelet aggregation
response (35% intensity of aggregation) was observed. The results shown in
Fig-1 manifest that optimal platelet aggregation is only recorded when
platelets are challenged with the subthreshold concentrations of epinephrine
and A23187 together. While decreasing the concentration of any one of the
agonist and keeping constant the concentration of the other agonist, does not
produce optimal platelet aggregation, thus establishing the synergistic effect
of epinephrine and A23187.
Fig-1a:
Percentage platelet aggregation responses induced by synergistic effect of
subthreshold concentration (0.5mM) of
A-23187 and different subthreshold concentrations (0.005, 0.01, 0.05, 0.1, 0.2mM) of Epinephrine
Fig-1b:
Percentage platelet aggregation responses induced by synergistic effect of
subthreshold concentrations (0.2mM) of
epinephrine and different subthreshold concentrations (0.6, 0.12, 0.25, 0.5 mM) of calcium ionophore A-23187.
When PRP was pretreated with different
concentrations, (0.01, 0.05, 0.1 μM) of Yohimbine an α2 adrenergic
receptor blocker and then the platelet aggregation was induced by adding
simultaneously the subthreshold concentration of epinephrine (0.2 μM) and
A23187 (0.5 μM), there was inhibition of platelet aggregation in a dose
dependent manner (Fig-2), IC50 value for the Yohimbine was calculated to be
0.05 μM. Similarly when the PRP was pretreated with different
concentrations (10,20,40,60 μM) of calcium channel blocker, Verapamil and
different concentrations (10,20,40,60,80,100 μM) of Diltiazem, another
calcium channel blocker, it inhibited
the platelet aggregation induced by subthreshold concentration of
epinephirine (0.2 μM) and subthreshold concentration (0.5 μM) of
A23187 in dose dependent manner . A dose response curve shown in Fig-3
manifests a dose dependent inhibitory effect of both the Verapamil and
Diltiazem. The IC50 values of Verapamil and Diltiazem as calculated from the
curve are 22 μM and 50 μM respectively. When PRP was pretreated with
different concentrations (0.1,0.2,0.4 μM) of SNAP, a nitric oxide (NO)
donor and then the platelet aggregation was induced by adding together the
subthreshold concentrations of epinephrine (0.2 μM) and A23187 (0.5
μM, the platelet aggregation was
inhibited in a dose dependent manner Fig-4. Dose response curve was constructed
and IC50 value was calculated to be
0.5μM.
Fig -2 Effect of different concentrations
(0.1, 0.05, 0.01mM) of Yohimbine (an a2 adrenergic receptor blocker) on platelet
aggregation induced by subthreshold concentrations of Epinephrine (0.2mM)
and A-23187 (0.5mM) shown as control
DISCUSSION
The results of the present study have shown that there
is potentiation of epinephrine effects by A23187 in human platelets. Our results also indicate that the platelet
activation induced by epinephrine is mediated through α2 adrenergic
receptors and ther is a definite role of Ca ++ in this
synergism. Furthermore the results also
exhibit that calcium channel blocking agents inhibit the platelet aggregation
induced as a result of epinephrine, A23187 synergism. While this synergism was also inhibited by
SNAP (a nitric oxide donor).
Fig-3: Dose response inhibitory effect of
verapamil and diltiazem on platelet aggregation induced by subthreshold
concentration of epinephrine (0.2mM) and A23187 (0.5mM).
Data is Mean ±SEM (n=5)
Fig-4: Effect of different concentrations
(0.4, 0.2, 0.1mM) of SNAP (nitric Oxide donor) on platelet
aggregation induced by subthreshold concentrations of epinephrine (0.2mM)
and A23187 (0.5 mM) shown as control
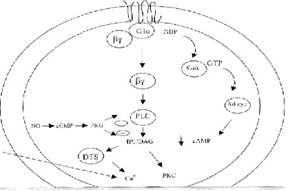
Fig -5: Proposed platelet model depicting the role of
Gi protein and calcium channels during coactivation by epinephrine and A-23187.
Gibg
subunit activates PLC, forming Inositol triphosphate (IP3) and
diacylglycerol (DAG).IP3 causing release of Ca++ from
dense tubular system (DTS) while A23187 promoting Ca++ entry into
the cytoplasm from outside. NO from exogenously added SNAP (NO donor) inhibits
platelet aggregation through production of cGMP. The cGMP activates protein
kinase G (PKG) which inhibits PLC induced IP3 formation.
In
our proposed platelet model, (Fig-5) epinephrine and A23187 synergism raises cytosllic Ca++
concentration on one hand by release from intracellular stores (dense tubular
system) and on the other hand by increased influx from the extracellular
fluid. Similar mechanism of agonist
syndergism is known among other agonists also and is considered to occur due to
activation of calcium signaling cascade.
Recent studies have shown that βχ subunits of activated Gi
protein can also activate phospholipase C (PLC)21-22.
Activation of PLC pathway leads to an increase in cytosolic Ca++
due to its release from internal stores i.e dense tubular system (DTS) by
inositol triphosphate (IP3) or through store depletd calcium influx23-24. The platelet cell membrane has limited
permeability to calcium but is penetrated by several channels capable of
permitting calcium influx. The calcium
channels are protein macromolecules.
Different types of calcium channels have been identified. Based on the stimulus required for opening of
channels, there are voltage dependent calcium channels (VDCC), receptor
operated channels (activated by binding of a chemical ligand to receptor) and
second messenger operated channels (cAMP dependent channels). Most of the calcium influx during platelet
activation results from passage through receptor operated calcium channels12. But the reagents that block VDCC, like Verapmil
and Diltiazem, can also prevent elevation of intracellular calcium induced by
several agonists 12-14.
To
study the molecular basis of this potentiation, SNAP, was used that inhibited
the platelet aggregation induced by the synergistic interaction of subthresthold
concentrations of epinephrine and A23187.
These results suggest that this synergism is sensitive to NO
generation. Platelets contain an
abundance of cAMP and cGMP dependent protein kinases which are activated by NO
and inhibit PLC induced IP3 and thramboxane receptors thus
inhibiting platelet aggregation24,25. The intracellular signaling involved in this
synergism is likely to be mediated through stimulation of PLC of βχ
subunits of Gi protein which in turn stimulates IP3 production and
thus mobilinze Ca ++ from intracellular stores in DTS. The DTS is rich in IP3 receptors
and Ca ++ is released whenever IP3 binds to its
receptor. Our results are in conformity
with the results of another study where the role of NO in inhibiting the
platelet aggregation has been claimed25. In another recent study [13] it has been
suggested that Gi and Gq proteins activation lead to PLC stimulation and Ca ++
signaling when the synergistic interaction of epinephrine and
5-hydroxytryptamine was studied. They
also observed the inhibitory effect of SNAP on this synergism. The results are similar to this study. The results of this study are in agreement
with another similar study18 where potentation of A23187 and
epinephrine has been shown. These
workers have also shown the inhibitory effect of calcium channel blocker,
Diltiazem. But these workers did not
explain intracellular signaling involved in this potentiation. In conclusion, our results reveal that
potentiation of epinephrine by A23187 involves raised cytosolic Ca++
concentration as a result of increased Ca ++ influx from ECF and
simultaneous mobilization of Ca++ from intracellular stores by
stimulation of PLC by βχ submit of Gi protein.
The
study also points to the communication between two different types of receptors
that exhibit synergism i.e. Gi protein linked α2 receptor and
calcium channels. In recent days, there
is growing evidence for such cross talk between different receptors that leads
to platelet aggregations 26-27
Acknowledgement
This study was supported by
REFERENCES
1.
Siess W. Molecular mechanism of platelet activation. Physiol Rev
1989;69:58-178.
2.
Grant JA, Scrutton MC. Novel α2 adrenoceptors primarily
responsible for inducing human platelet aggregation. Nature 1979;277:659-61.
3.
Homcy CJ. Garham RM. Molecular characterization of adrenergic receptors.
Circ Res 1985:635-650.
4.
Aktories K, Jakobs H. Eipnephrine inhibits adenylate cyclase and
stimulates a GTPase in human platelet membrances via alpha-adrenoceptors. FEBS
lett 1981;130: 235-38.
5.
Connolly TM, Limibird LE. The influence of Na+ on the alpha
adrenergic receptor system of human
platelets. A method for removal of
extraplatelet Na+: Effects of Na+ removal on aggregation
secretion and cAMP accumulation. J Biol Chem 1983;258:3907-12.
6.
Steen VM, Tysnes OB, Holmsen H. Synergism between thrombin and
adrenaline (epinephrine) in human platelets Marked potentitation of
inositol phospholipids metabolism. Biochem J 1988;253:581-6.
7.
Steen VM,
8.
Banga HS, Somins ER, Brass LF, Rittenhouse SE. Activation of
phospholipase A and C in human platelets exposed to epinephrine: role of
glycoprotein II b/III a and dual role of epinephrine. Proc Natl Acad Aci
9.
Steen VM, Holmsen H, Aarbakke G. The platelet stimulating effect of
adrenaline through agonist. Thromb Haemost 1993;70(3):506-13.
10. Razi MS, Butt IF, Ayub M,
Aslam M, Hameed W, Badar A, et al. Synergistic Interaction of Adenosine
diphosphate-Epinephrine and Epinephrine- Collagen in aggregation of Human
Platelets. J Ayub Med Coll Abbottabad 2004;16(3):20-24.
11. Ware JA, Smith M, Salzman EW. Synergism
of platelet aggregating agents: Role of elevation of cytoplasmic calcium. J
Clin Invest 1987; 80:267-71.
12. Shah BH, Siddiqui A, Qureshi
KA, Khan M, Rafi S, Ujan VA et al.
Coactivation of Gi and Gq proteins exerts synergistic effect on human
platelet aggregation through activation of phosphlipase c and Ca ++ signaling
pathways. Exp Mol Med
1999;31:42-46.
13. Grant J A, Scrutton MC.
Positive interaction between agonists in the aggregation response of human blood
platelets: Interaction between ADP,
adrenaline and vasopressin. Br J
Haematol 1980;44:109-125.
14. Steen VM, Holmsen H. Synergism
between thrombin and epinephrine in human platelets: Different dose-response
relationship for aggregation and dense granule secretion. Thromb Haemost 1985; 30:680-9.
15. Packham MA, Guccione MA, Chang
PL, Mustard JF. Platelet aggregation and release; effects of low
concentrations of thrombin or collagen.
Am J Physiol 1973; 225:38-47.
16. Figures WR, Scearce LM,
Wachtfogel Y, Chen J, Colman RF, Colman RW.
Platelet ADP receptor and α2 adrenorceptor
interaction. J Biol Chen 1986;
261:5981-5986.
17. Sation M, Salzman EW, Smith H,
Ware JA. Activation of ptorein kinase C
in platelets by epinephrine and A23187;
Correlation with fibrinogen. Blood 1989;74:2001-7.
18. Kinlough-Rathbone RL, Mustard
JF. Synergism of agonists. In Platelet Response and Metabolism. Holmser H;
editor CRB Press, Boca Ration E.L 1986;193-207.
19. Born GVR. Adenosine
diphosphate as a mediator of platelet aggregation in vivo; an editorial view.
Circulation 1985;72(4):741-6.
20.
21. Banno Y, Asano T, Nozawa
Y. Stimulation by G Protein βγ
sub units of phopholipase C isoforms in human platelets. Thromb Haemost
1998;79:1008-1013.
22. Berridge MJ. Inositol
trisphosphate and calcium signaling Nature 1993;361: 315-325.
23. Hemskerk JWM, Sage O. Calcium
signaling in platelets and other cells.
Platelets 1994;5:295-316.
24. Wang GR, Zhu Y, Haluskha PV,
Lincoln TM, Mendelsohn ME. Mechanism of
platelet inhibiton by nitric oxide: in vivo phosphorylation of thromboxane
receptor by cyclic GMP-dependent protein kinase. Proc Natl Acad Sci
25. Wang X, Yanagi S, Yang C,
Inatome R, Yamunura H. Tyrosine
phosphorylation and SYK activation are involved in thrombin induced aggregation
of epinephrine-potentiated platelets.
JBiochemist 1997;121:322-330.
26. Nieswandt B, Bergmeier W,
Eckly A, Schutle V, Ohlmann P, Cazenave JP, Ziringibl H et al. Evidence for cross cross talk between
gloycoprotein VI and Gi coupled receptors during collagen induced platelet
aggregation. Blood 2001;97 (12); 3829-3825.
27. Butt I F, Saeed S A, Rasheed
H, Aslam M. Role of Phosphatidyl Inositol 3 Kinase in Epinephrine and Calcium
Iononophore A 23187 induced Platelet Aggregation. Pak Armed Forces Med J 2004;
54 (1): 86-91.